Whole-genome sequencing (WGS) is widely used to study human genomics; however, non-human nucleic acids can sometimes show up in human WGS data. When viruses, for example, are present in the sample, high throughput sequencing can capture traces of their nucleic acids alongside those of humans, and therefore they can sometimes be detected, “hidden” within human WGS datasets.
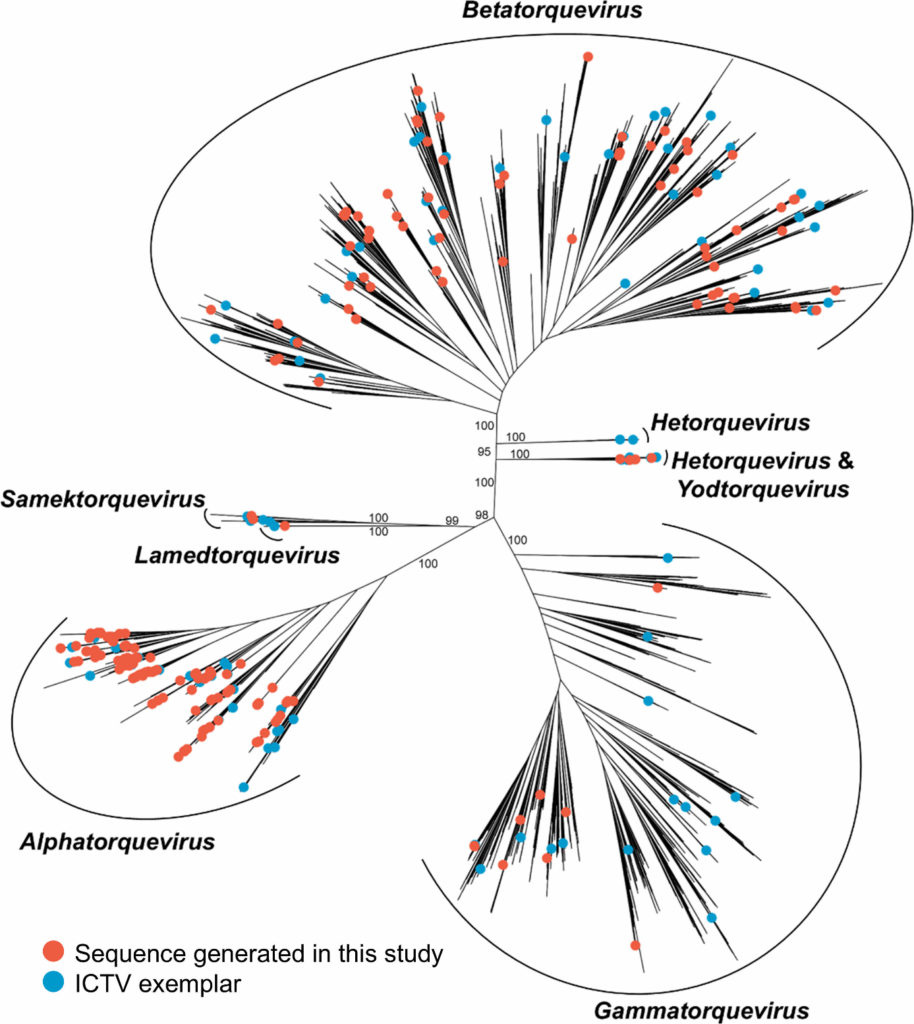
In a new study published in Microbiology Spectrum, researchers analysed 1,175 WGS datasets from Thai individuals using Entourage, a virus-mining pipeline, and uncovered hundreds of anellovirus sequences. With 434 partial genomes and the first 77 complete genome sequences of Thai anelloviruses, this study reports the largest and most well-curated collection of anellovirus sequences reported from Thailand to date.
Until now, only three anellovirus genera have been reported in Thai human individuals (Alphatorquevirus, Betatorquevirus, and Gammatorquevirus). This study expands the landscape of known Thai anelloviruses in humans to include four more genera (Hetorquevirus, Lamedtorquevirus, Samektorquevirus, and Yodtorquevirus). In addition, the researchers identified 33 potentially novel species within the genera Alphatorquevirus (six potentially novel species), Betatorquevirus (23 potentially novel species), and Gammatorquevirus (four potentially novel species). Their analyses also suggest frequent cross-border transmission of anelloviruses between Thailand and other countries. The authors hope that their results will help establish a foundation for future anellovirus studies in Thailand.
The study also examined the virus current species classification system, which relies mainly on pairwise similarity analysis of complete orf1 sequences—the largest gene encoding the virus capsid protein. While pairwise similarity analysis often gives results consistent with virus evolutionary relationships, the authors found that this was not always the case—particularly when sequence alignments contain many gaps. In such cases, relying solely on similarity measures could incorrectly group very distantly related viruses together. They recommend that similarity-based classification should be cross-checked with phylogenetic analysis to ensure consistency with evolutionary history, especially when the similarity values are calculated from alignments with high gap proportions.
While mining human WGS for viral sequences is not yet a routine practice, the study highlights its potential for virus discovery. With sequencing technologies and tools for sequence analysis becoming increasingly accessible worldwide, we can expect to see new anelloviruses reported from underexplored regions and populations, and this will undoubtedly reshape our understanding of anellovirus diversity and taxonomy.
First Author: Worakorn Phumiphanjarphak
Corresponding Author: Pakorn Aiewsakun